
Синдром Ретта


Синдром Ретта: общие сведения и история открытия
Синдром Ретта — редкое врождённое заболевание нервной системы, которое в большинстве случаев обнаруживается у девочек.
В первые месяцы жизни ребёнок, как правило, развивается нормально: учится держать голову, сидеть, может играть и произносить отдельные звуки. Затем, чаще всего в возрасте от шести месяцев до полутора лет, развитие замедляется, а уже приобретённые навыки начинают утрачиваться.
С возрастом у девочек с синдромом Ретта могут появиться проблемы с движением — неустойчивая походка, скованность мышц, выраженная слабость. У многих возникают судороги, неровное дыхание, проблемы с пищеварением и набором веса. Речь исчезает. Останавливается рост головы, и она начинает казаться слишком маленькой по отношению к телу.
Название болезни происходит от фамилии врача, который первым подробно описал синдром.

Андреас Ретт — австрийский педиатр, невролог и писатель, который посвятил свою жизнь лечению детей с нервными расстройствами. Автор: Media Wien, CC BY-SA 3.0, via Wikimedia Commons
В 1950–60-е годы австрийский педиатр и невролог Андреас Ретт работал в Вене с детьми, страдавшими тяжёлыми нервными расстройствами. Он обратил внимание, что у нескольких девочек заболевание протекает сходным образом: сначала дети развиваются нормально, затем речь и навыки пропадают. Появляются характерные повторяющиеся движения, напоминающие мытьё рук.
В 1966 году Ретт опубликовал свои наблюдения в немецкоязычном медицинском журнале, но его работа долгое время оставалась неизвестной за пределами немецкоговорящих стран. Лишь в начале 1980-х годов, когда шведский врач Бенгт Хагберг и его коллеги описали аналогичные случаи у девочек и опубликовали результаты в англоязычном журнале, болезнь получила международное признание.
С этого момента исследователи стали активно изучать синдром Ретта, его симптомы, стадии, отличия от аутизма и других подобных патологий.
По разным оценкам, синдром Ретта обнаруживается у одной из 10–15 тыс. новорождённых девочек. Риск заболеть не зависит от расы, этнической принадлежности, региона проживания.
и делаем скидки до 30%
Начните экономить прямо сейчас!
Причины синдрома Ретта
Основная причина синдрома Ретта — мутация гена MECP2, который находится на X-хромосоме.
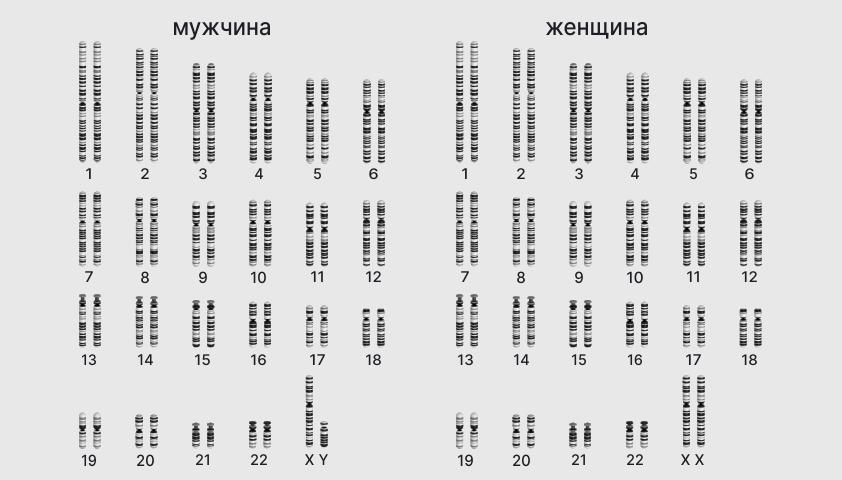
Хромосомный набор человека: 22 пары хромосом, которые содержат «инструкции» по развитию организма, и одна пара, отвечающая за пол (XY или XX)
Ген MECP2 отвечает за нормальную работу нервных клеток — помогает «включать и выключать» другие гены в нужный момент, чтобы головной мозг правильно развивался и функционировал. Если MECP2 «сломан», то нервные клетки действуют несогласованно, что приводит к проблемам с речью, дыханием, движениями и поведением.
Синдром Ретта почти всегда обнаруживается у девочек, поскольку у них есть две X-хромосомы. Если одна из них содержит повреждённый ген MECP2, то вторая частично компенсирует его работу. Поэтому ребёнок выживает, но он болен.
У мальчиков имеется только одна X-хромосома, поэтому такая же мутация у них обычно несовместима с жизнью.
В большинстве случаев генетическая мутация возникает спонтанно, однако известны факторы риска, способные её спровоцировать, — краснуха во время беременности, приём беременной женщиной сильнодействующих лекарственных препаратов, а также слишком частые (с перерывом менее 1 года) беременности.
Семейные случаи синдрома Ретта встречаются крайне редко: если в семье уже родился ребёнок с таким диагнозом, это не означает, что следующий также будет болен.
В редких случаях синдром Ретта (часто атипичный) вызывают мутации в других генах, таких как CDKL5 или FOXG1.
Виды синдрома Ретта
Наиболее известна и изучена классическая форма синдрома Ретта. Ребёнок с таким диагнозом развивается нормально в течение первых 6–18 месяцев жизни, затем прогресс останавливается: речь исчезает, появляются характерные стереотипные движения кистей — будто малыш моет руки. Со временем ребёнку становится трудно ходить, дышать.
Есть также атипичные формы синдрома Ретта. Они получили имена исследователей, которые первыми подробно описали их.
Атипичные формы синдрома Ретта:
- вариант Заппеллы (Zappella): у ребёнка частично сохраняется речь и способность к осмысленному общению. Девочки могут произносить отдельные слова и фразы. Физическое развитие близко к возрастной норме;
- вариант Ханефельда (Hanefeld): у ребёнка очень рано возникают судорожные приступы. Эпилепсия становится основным симптомом болезни;
- вариант Роландо (Rolando): на первый план выходят трудности с сидением, ходьбой, координацией движений. Часто наблюдается неустойчивая походка, слабость мышц.
Стадии и симптомы синдрома Ретта
Синдром Ретта развивается последовательно: стадии болезни сменяют друг друга по мере роста и развития ребёнка. И каждая из них сопровождается определённым набором симптомов со стороны нервной системы, внутренних органов, а также особенностями поведения.
Осложнения синдрома Ретта
Чаще всего у детей с синдромом Ретта возникают проблемы со стороны нервной системы. У многих развиваются судороги — от кратких эпизодов «замирания» до выраженных эпилептических приступов.
Со временем также появляются трудности с движением: ребёнку становится трудно ходить, удерживать равновесие. Движения могут становиться скованными.

Из-за проблем с равновесием, мышечной слабости и искривления позвоночника многие девочки теряют способность ходить. Автор: Senorita666, CC BY-SA 4.0, via Wikimedia Commons
Для синдрома Ретта характерно нарушение дыхания, особенно когда ребёнок бодрствует. Это могут быть задержки, частое или нерегулярное дыхание, которое сопровождается криком или беспокойством.
С питанием и пищеварением также часто возникают сложности. Многим детям сложно жевать и глотать, появляется сильное слюнотечение, изжога, запоры. Из-за этого ребёнок может плохо набирать вес, нередко развивается дефицит витаминов и минералов.
У некоторых детей нарушается работа сердца и вегетативной нервной системы (она отвечает за дыхание, сердцебиение, пищеварение, работу сосудов). Характерные признаки — холодные, иногда синюшные руки и ноги, зябкость или непереносимость тепла, изменения сердечного ритма.
Диагностика синдрома Ретта
Диагностировать синдром Ретта бывает непросто, поскольку эта болезнь начинается не с одного яркого симптома, а с постепенных изменений в развитии детей.
Как правило, родители или педиатр замечают, что ребёнок перестал интересоваться игрушками, указывать на людей и предметы. Вместо этого у него появились характерные стереотипные движения рук — «мытьё», хлопанье, заламывание, постукивание, скручивание, потирание. При этом он не произносит отдельные звуки или уже усвоенные слова.
Если появились подобные признаки (до 6–18 месяцев нормальное развитие → потеря навыков → характерные движения рук), следует как можно скорее обратиться к неврологу.
На приёме врач уточнит, перенесла ли женщина краснуху во время вынашивания ребёнка, принимала ли сильнодействующие лекарственные препараты, как прошли роды.
Затем невролог перейдёт к осмотру: обратит внимание на движения рук ребёнка, проверит, есть ли трудности с дыханием, координацией движений. Также он обязательно измерит окружность головы и проверит, соответствует ли этот показатель возрастным нормам.
Следующий этап — генетическое тестирование. У девочек с синдромом Ретта обнаруживается мутация в гене MECP2, расположенном на X-хромосоме.
Если симптомы болезни похожи на синдром Ретта, но мутация MECP2 не обнаружена, может потребоваться расширенная генетическая диагностика, которая поможет выявить другие заболевания со сходными проявлениями, например синдром Ангельмана, Питта — Хопкинса или Корнелии де Ланге.
Дополнительно проводят инструментальные исследования — электроэнцефалографию (ЭЭГ), магнитно-резонансную томографию (МРТ) головного мозга, электрокардиографию (ЭКГ) сердца. Все они помогают оценить состояние здоровья ребёнка и определить риск осложнений.
При подозрении на наследственную форму синдрома Ретта врач может рекомендовать консультацию у генетика.
Лечение синдрома Ретта
Специального лечения синдрома Ретта не существует. Терапия направлена на то, чтобы облегчить состояние ребёнка и предупредить осложнения.
При судорогах, частых эпилептических припадках назначают противоэпилептические препараты. Для того чтобы сохранить способность стоять или ходить как можно дольше, показаны занятия лечебной физкультурой.
Если ребёнку трудно самостоятельно принимать пищу, врач может назначить лечебное питание — нутритивные смеси, которые помогают обеспечить организм всеми необходимыми для жизни веществами. Такие смеси содержат сбалансированное количество белков, жиров, углеводов, витаминов и минералов и используются при трудностях с глотанием, жеванием.
Большое значение имеет работа с коммуникацией. Хотя речь у большинства детей с синдромом Ретта утрачена или сильно ограничена, многие из них способны понимать простые слова и выражать эмоции. Логопеды и специалисты по альтернативной коммуникации помогают подобрать способы общения через взгляды, жесты, карточки, планшеты. Это не обучение речи, а возможность дать ребёнку способ сообщить о своих потребностях и быть услышанным.
Если у ребёнка есть выраженные нарушения в работе сердца или трудности с дыханием, могут потребоваться лекарственные препараты и регулярные ЭКГ.
Прогноз и профилактика синдрома Ретта
Прогноз синдрома Ретта во многом зависит от формы болезни и выраженности осложнений.
При хорошем уходе и регулярной поддержке со стороны врачей многие девочки с таким диагнозом могут дожить до 20–30 лет. Но большинство из них способны передвигаться только на инвалидной коляске.
Специфической профилактики синдрома Ретта нет. Однако следует обязательно учитывать факторы, повышающие риск развития болезни.
На этапе планирования беременности будущим родителям полезно исследовать свой хромосомный набор, чтобы исключить патологии, которые могут передаться ребёнку. Во время беременности — избегать приёма лекарственных и наркотических препаратов, регулярно посещать врача, а также учитывать, что вероятность генетических аномалий с возрастом повышается: в группе риска — женщины, которым на момент зачатия ребёнка было более 40 лет.
Источники
- Ворсанова С. Г., Юров Ю. Б., Воинова В. Ю. и др. Синдром Ретта в России и за рубежом: научный исторический обзор // Российский вестник перинатологии и педиатрии. 2020. №2. С. 25–31.
- Лаптева Н. М., Маркова О. М., Алексеева Т. С. и др. О случае орфанного заболевания — синдром Ретта в практике педиатра // Наука и мир. 2016. №8 (36). С. 69–71.
- Horvath L. MeCP2 as an activator of gene expression // Trends in Neurosciences. 2018. Vol. 41(2). P. 72–74.
Частые вопросы
врач-эксперт



