


и делаем скидки до 30%
Начните экономить прямо сейчас!
Что такое мукополисахаридоз
Мукополисахаридоз — группа редких генетических заболеваний, при которых в соединительной ткани накапливается большое количество мукополисахаридов (или гликозаминогликанов) — сложных молекул сахара.
Соединительная ткань — связующее звено, которое объединяет все ткани и органы и формирует их каркас (в медицине его называют стромой). Кроме того, именно из соединительной ткани состоит дерма — средний слой кожи человека.
Примерно 50% веса человека приходится на соединительную ткань.
Обнаружить соединительную ткань можно в кровеносных сосудах и нервах, хрящах и костях, мышцах и связках. Также она присутствует в жировой ткани, лимфе, подкожной клетчатке и даже радужке глаза.

Так выглядит соединительная ткань под микроскопом
Один из основных компонентов соединительной ткани — протеогликаны. Это сложные химические соединения, которые на 5–10% состоят из белка и на 90–95% из длинных цепочек молекул сахара — гликозаминогликанов (другое название — мукополисахариды).
В норме протеогликаны живут всего 7–10 дней, затем под действием специальных ферментов распадаются.
При мукополисахаридозе в организме отсутствуют или не функционируют как следует ферменты, способные разрушить протеогликаны. В результате гликозаминогликаны (ГАГ) начинают откладываться в соединительной ткани.
Сами по себе ГАГ нетоксичны, но если в организме накапливается слишком большое их количество, то от этого начинают страдать различные ткани и органы. Развивается мукополисахаридоз.
В основном заболевание наследуется по аутосомно-рецессивному типу. Это значит, что болезнь ребёнку передают здоровые родители — носители дефектного гена.
Если ген болезни есть только у одного из родителей, то мукополисахаридоз у ребёнка не возникнет, так как здоровый ген второго родителя сможет компенсировать ситуацию: организм будет синтезировать 50% от необходимого уровня ферментов и этого, вероятнее всего, окажется достаточно, чтобы заболевание не развилось. Но если ген болезни имеется и у отца, и у матери, то с вероятностью 25% ребёнок получит диагноз «мукополисахаридоз».
Исключение из правила — мукополисахаридоз II типа. Он наследуется по X-сцепленному рецессивному типу.
Такой вариант мукополисахаридоза в абсолютном большинстве обнаруживается у мальчиков, так как у них есть только одна X-хромосома, а не две, как у девочек. Соответственно, чтобы мальчик заболел, достаточно, чтобы он унаследовал только одну копию дефектного гена.

Мукополисахаридоз — редкое заболевание. Оно обнаруживается примерно у одного из 140 000 новорождённых
Виды и симптомы мукополисахаридоза
Науке известно 11 типов мукополисахаридоза, которые различаются между собой степенью тяжести болезни. Кроме того, у отдельных типов может быть несколько разновидностей.
Мукополисахаридоз типа I-H
Мукополисахаридоз типа I-H (синдром Гурлер) — один из самых распространённых видов этой болезни.
Синдром Гурлер наследуется по аутосомно-рецессивному типу и, как правило, проявляется уже с первых месяцев жизни ребёнка.
Основные симптомы мукополисахаридоза типа I-H:
- умственная отсталость;
- задержка психомоторного развития (двигательных и интеллектуальных навыков);
- задержка роста (обычно дети дорастают до 110 см, после этого рост полностью останавливается);
- кифоз (искривление) поясничного отдела позвоночника;
- пупочные или паховые грыжи;
- грубые черты лица;
- тугоподвижность суставов;
- помутнение роговицы глаза;
- диарея в раннем возрасте;
- тугоухость;
- плохое зрение.

Характерные внешние признаки синдрома Гурлер — крупный череп, выступающий лоб, широкие скулы, запавшая переносица, толстые губы, короткая шея
Как правило, дети с таким диагнозом погибают в возрасте примерно 10 лет от сердечной недостаточности, респираторных инфекций или обструкции дыхательных путейОбструкция дыхательных путейНарушение проходимости дыхательных путей, при котором воздух перестаёт поступать в лёгкие..
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S (синдром Шейе) — более лёгкая форма заболевания.
Первые симптомы болезни проявляются к возрасту 3–5 лет — до этого времени развитие соответствует норме. Затем у ребёнка возникают сложности со сгибанием пальцев рук, запястья, локтей и плеч. Суставы ног при этом, как правило, не страдают.
Основные симптомы мукополисахаридоза типа I-S:
- тугоподвижность суставов;
- низкорослость;
- неуклюжесть;
- грубые черты лица;
- помутнение роговицы глаза;
- непропорционально крупный череп;
- недоразвитие одной или обеих челюстей;
- умеренный гипертрихоз — повышенное оволосение;
- кифоз (искривление) поясничного отдела позвоночника;
- пупочные или паховые грыжи;
- X-образные голени;
- иногда — незначительное отставание умственного развития.

Помутнение роговицы глаза — один из симптомов мукополисахаридоза
Продолжительность жизни людей с синдромом Шейе может достигать 30 лет.
Мукополисахаридоз II типа
Мукополисахаридоз II типа (синдром Хантера) — второй по распространённости тип болезни после синдрома Гурлер.
Синдром Хантера — единственный тип мукополисахаридоза, наследуемый по X-сцепленному рецессивному типу. В подавляющем большинстве случаев он развивается у мальчиков. А девочки становятся бессимптомными носителями болезни.
В зависимости от формы синдрома Хантера, первые симптомы могут появиться в возрасте 1,5–3 лет (тяжёлая форма — IIA) или в 3–8 лет (среднетяжёлая форма — IIB).
Пациенты с синдромом Хантера IIA, как правило, погибают на втором десятилетии жизни. Люди с формой IIB могут прожить 30–40 лет.
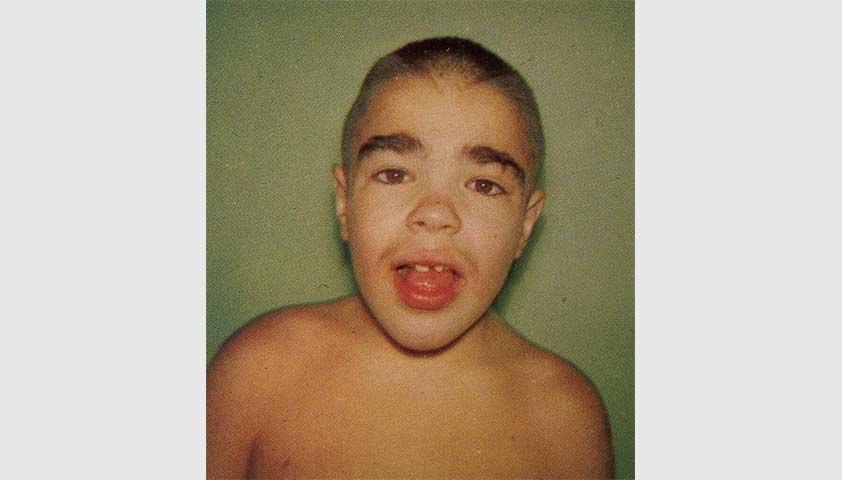
На фото — ребёнок с синдромом Хантера. Сейчас в мире насчитывается около 2 000 людей с такой болезнью, в России — около 100
Основные симптомы мукополисахаридоза II типа:
- тугоподвижность суставов;
- низкорослость;
- грубые черты лица;
- непропорционально крупный череп;
- пупочные и паховые грыжи;
- аномалия зубных рядов (слишком редкие зубы);
- шумное дыхание;
- хриплый низкий голос;
- частые простуды, отиты;
- хроническая диарея;
- умеренный гипертрихоз — повышенное оволосение;
- помутнение роговицы глаза;
- тугоухость;
- задержка речевого развития.
Мукополисахаридоз III типа
Мукополисахаридоз III типа (синдром Санфилиппо) отличается от других типов тем, что гликозаминогликаны (ГАГ — длинные цепочки молекул сахара) скапливаются в основном в головном и спинном мозге.
Первые тревожные звоночки, которые могут указывать на болезнь, появляются ещё на этапе беременности. У женщин, которые вынашивают ребёнка с синдромом Санфилиппо, часто возникает угроза самопроизвольного выкидыша. При родах нередко возникает слабость родовой деятельности — в этом случае может потребоваться экстренное кесарево сечение.
Сразу после рождения у детей развивается затяжная желтуха.

При желтухе в крови повышается уровень пигмента билирубина. Кожа и слизистые оболочки младенца окрашиваются в жёлто-оранжевый цвет
Типичные для синдрома Санфилиппо симптомы появляются на втором году жизни ребёнка.
Основные симптомы мукополисахаридоза III типа:
- лёгкое огрубение черт лица;
- незначительная тугоподвижность суставов;
- низкорослость;
- утрата моторных (двигательных) навыков и способности связно говорить;
- гиперактивность;
- истеричное поведение;
- агрессивность;
- нарушение сна.

Дети с синдромом Санфилиппо внешне могут почти не отличаться от сверстников. Автор: Noamorv, CC BY-SA 4.0, via Wikimedia Commons
По мере прогрессирования болезни дети становятся всё менее подвижными и активными. В некоторых случаях им требуется инвалидная коляска.
Продолжительность жизни пациентов с синдромом Санфилиппо около 20 лет.
Мукополисахаридоз IV типа
Мукополисахаридоз IV типа (синдром Моркио) поражает в основном кости и суставы. Интеллект, как правило, не страдает.
Первые симптомы появляются в возрасте 1–3 лет, а к 7–8 годам жизни они становятся наиболее яркими.
Основные симптомы мукополисахаридоза IV типа:
- задержка физического развития;
- низкорослость;
- аномалия зубных рядов (слишком редкие зубы);
- слабость мышц;
- искривление грудного и поясничного отдела позвоночника;
- тугоподвижность суставов;
- тугоухость;
- пупочные грыжи.

Дети с синдромом Моркио кажутся здоровыми при рождении. Болезнь манифестирует в возрасте 1–3 лет
Продолжительность жизни людей с диагнозом «синдром Моркио» — 20–35 лет.
Другие типы мукополисахаридоза
Мукополисахаридозом V типа раньше считали синдром Шейе. Сейчас его относят к I типу (лёгкой форме синдрома Гурлер).
Мукополисахаридоз VI типа (синдром Марото — Лами) обычно манифестирует в возрасте 2 лет. Основные симптомы — грубые черты лица, низкорослость, короткая шея, тугоподвижность суставов, бочкообразная деформация грудной клетки. Интеллект при этом, как правило, не страдает.
Продолжительность жизни людей с синдромом Марото — Лами приблизительно 20 лет.
Мукополисахаридоз VII типа (синдром Слая) протекает так же, как и синдром Санфилиппо. Страдают преимущественно головной и спинной мозг. Продолжительность жизни при этом составляет около 20 лет.
Осложнения мукополисахаридоза
Средняя продолжительность жизни людей с диагнозом — менее 20 лет. При этом пациенты погибают не от самого заболевания, а от его осложнений.
Возможные осложнения мукополисахаридоза:
- тяжёлые рецидивирующие инфекции (отиты, риниты, синуситы), которые могут привести к дыхательной недостаточности;
- нарушение работы печени и селезёнки;
- серьёзные деформации скелета и, как следствие, невозможность ходить;
- болезни сердца: миокардиодистрофия (истончение сердечной мышцы — миокарда), стеноз (сужение) сердечных клапанов, гипертрофия левого желудочка (утолщение стенки желудочка, которое может привести к перебоям в работе сердца, а затем и к смерти);
- лёгочное сердце — расширение правых отделов органа, при котором сердце не может работать как следует;
- необратимое повреждение головного мозга;
- стеноз (сужение) позвоночного канала и, как следствие, передавливание спинного мозга.
К какому врачу обращаться при симптомах мукополисахаридоза
При подозрении на мукополисахаридоз ребёнка нужно как можно быстрее показать педиатру. В ходе обследования пациенту потребуются консультации других специалистов — генетика, кардиолога, офтальмолога, гастроэнтеролога, оториноларинголога (лора), психиатра, ортопеда.
Лабораторная диагностика
Базовое исследование, которое позволяет выявить мукополисахаридоз, — анализ крови или мочи на гликозаминогликаны. Исследование полезно для раннего скрининга болезни, но неспособно определить конкретный тип мукополисахаридоза. С этой целью пациенту назначают генетический анализ.
Всем парам, которые планируют беременность, целесообразно сдать анализ на носительство наиболее распространённых генетических мутаций. Результаты исследования помогут понять, каков риск рождения ребёнка с тяжёлой наследственной болезнью.
Инструментальная диагностика
Инструментальные исследования позволяют оценить масштаб поражений внутренних органов и систем.
Исследования, которые могут назначить при подозрении на мукополисахаридоз:
- УЗИ, КТ (компьютерная томография) или МРТ (магнитно-резонансная томография) органов брюшной полости;
- рентгенография шейного, грудного, поясничного отдела позвоночника, тазобедренных суставов, рук и ног (при мукополисахаридозе на снимках будут видны деформации костей, искривление позвоночного столба);
- электронейромиография (ЭНМГ) — оценка состояния спинного мозга и мышечных волокон;
- эндоскопическая эндоназальная ревизия полости носа, околоносовых пазух и носоглотки (у пациентов с мукополисахаридозом уже с раннего возраста обнаруживается разрастание миндалин);
- аудиометрия — измерение остроты слуха;
- спирометрия — оценка дыхательной функции;
- электроэнцефалография (ЭЭГ) — оценка состояния нервной системы;
- полисомнография — исследование нарушений сна;
- электрокардиография (ЭКГ) — оценка работы сердца;
- офтальмоскопия — осмотр глазного дна.
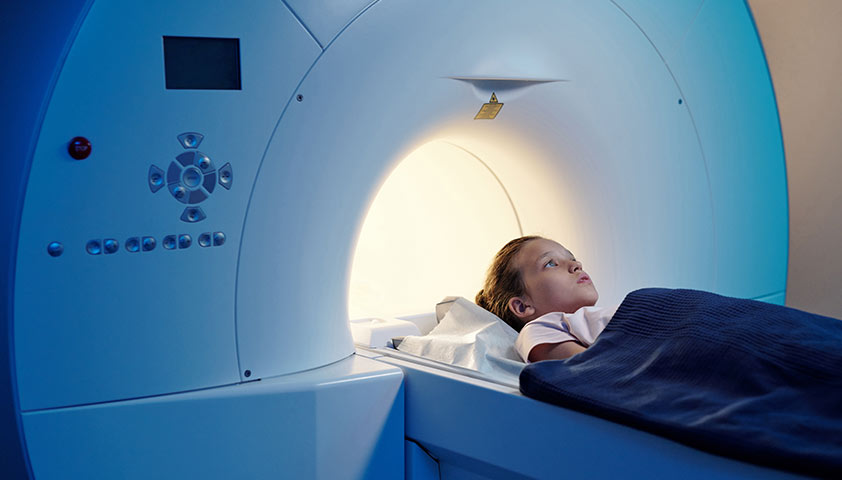
Магнитно-резонансная томография (МРТ) позволяет получить детальное изображение органов и тканей
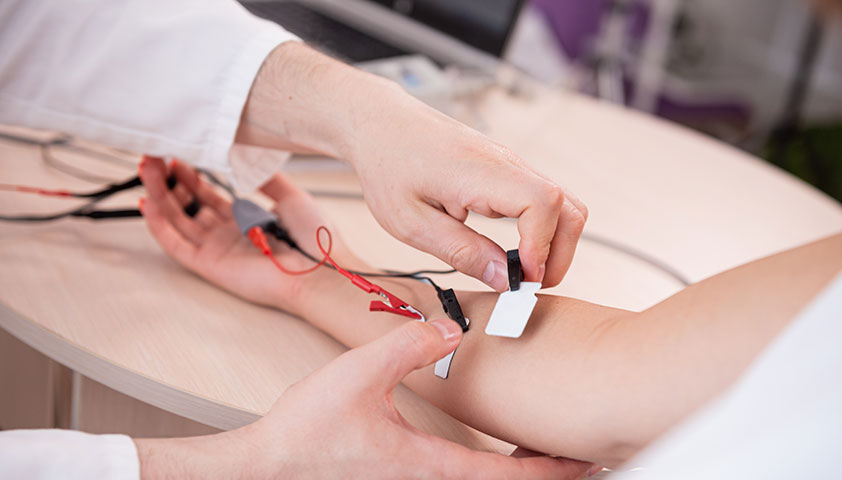
С помощью электронейромиографии (воздействия током на мышцы и нервные окончания) можно оценить состояние спинного мозга и мышечных волокон

Аудиометрия — один из аппаратных способов диагностики тугоухости
Лечение мукополисахаридоза
Лечению поддаются только три разновидности мукополисахаридоза — I, II и VI типы. Пациентам с такими диагнозами назначают лекарственные препараты, которые позволяют заместить недостающие ферменты.
В России зарегистрированы три препарата: ларонидаза (используется для лечения мукополисахаридоза I типа), идурсульфаза (назначается при II типе) и галсульфаза (показана при VI типе).
Принимать синтетические ферменты пациентам с диагнозом «мукополисахаридоз» придётся пожизненно.
Некоторым пациентам выполняют трансплантацию костного мозга. Такая операция призвана заставить организм вырабатывать собственные ферменты, необходимые для переработки гликозаминогликанов.
Наилучшие результаты были получены при пересадке костного мозга детям до 2 лет с диагнозом «мукополисахаридоз I типа». У пациентов с мукополисахаридозом II типа трансплантация может привести к серьёзным осложнениям и даже к смерти.
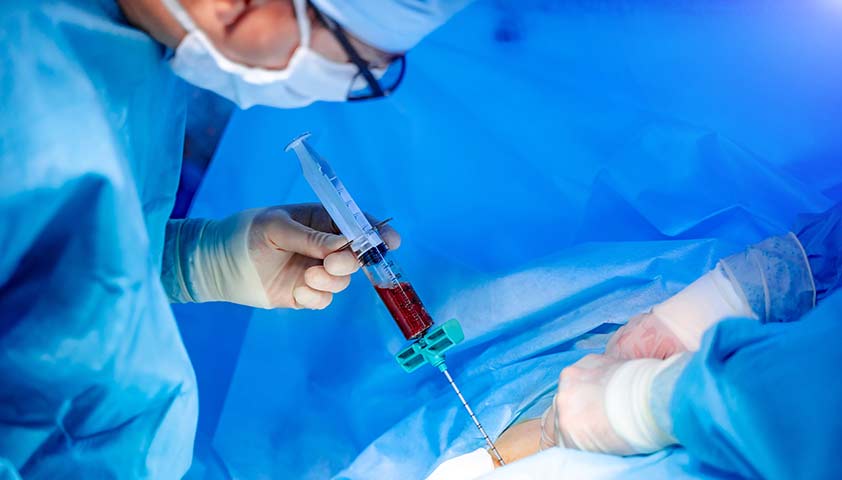
Костный мозг для трансплантации получают от подходящего здорового человека. Для донора процедура взятия клеток безопасна и практически безболезненна
В последние годы генетики активно работают над созданием лекарства, которое помогло бы пациентам с мукополисахаридозом. Учёные пытаются создать вирусные векторыВирусные векторыОбезвреженные вирусные частицы, которые используются для доставки генетического материала внутрь клеток организма., которые смогли бы доставить в организм нужный ген и встроить его в заданный участок хромосомы вместо дефектного гена.
Симптоматическое лечение мукополисахаридоза направлено на улучшение самочувствия пациента.
Лекарственные препараты, которые применяют для симптоматической терапии:
- антибиотики (для лечения тяжёлых рецидивирующих инфекций);
- транквилизаторы, седативные и ноотропные препараты (для коррекции поведенческих нарушений);
- антигипертензивные препараты, разрешённые для детей (при высоком артериальном давлении).
Многим пациентам требуется коррекция осанки. В большинстве случаев для этого используют ортезы — устройства, которые помогают разгрузить мышцы и поддерживают анатомически правильное положение костей и суставов.
При мукополисахаридозе I и VI типа также может потребоваться операция, призванная остановить прогрессирующее искривление позвоночника. После операции пациенту придётся провести в корсете 3–6 месяцев.
При мукополисахаридозе у пациентов часто развивается карпальный туннельный синдром — заболевание, при котором нерв, отвечающий за движение кисти, сдавливается и воспаляется. Чтобы сохранить подвижность руки, пациенту могут назначить операцию для освобождения сдавленного нерва.
Прогноз и профилактика
Прогноз зависит от типа мукополисахаридоза. В большинстве случаев заболевание приводит к инвалидности, а затем и к смерти пациента.
Дети с синдромом Гурлер (тип I-H) без лечения погибают в возрасте 10 лет. Пациенты с синдромом Шейе (более мягкая форма болезни, тип I-S) могут дожить до 30 лет. Люди с синдромом Санфилиппо (тип III) иногда доживают до 20 лет.
При лечении в некоторых случаях пациентам удаётся преодолеть такие рубежи и их продолжительность жизни становится чуть выше.
Специфических способов профилактики нет. Единственная возможность предупредить болезнь — пройти исследование на носительство генетических мутаций, которые могут привести к тяжёлой болезни у ребёнка. Сдать такой анализ целесообразно всем парам, которые планируют стать родителями.
Источники
- Федеральные клинические рекомендации по диагностике и лечению мукополисахаридоза I типа / Министерство здравоохранения РФ. 2013.
- Hamosh A. Hurler syndrome. Mucopolysaccharidosis type IH; MPS1-H // Online Mendelian Inheritance in Man. 2018.
- Demczko M. Mucopolysaccharidoses // MSD Manuals. 2018.
- Семячкина А. Н., Новиков П. В., Воскобоева Е. Ю. и др. Мукополисахаридозы у детей // Российский вестник перинатологии и педиатрии. 2007. № 4. С. 22–29.
- Моисеев С. В., Новиков П. И., Фомин В. В. Мукополисахаридозы — путь к диагнозу // Клиническая фармакология и терапия. 2018. № 3. С. 41–47.
Частые вопросы
Мукополисахаридоз — это группа редких (выявляют примерно у одного из 140 000 новорождённых) генетических заболеваний, при которых в соединительной ткани накапливается большое количество мукополисахаридов (или гликозаминогликанов) — сложных молекул сахара. От этого страдают различные ткани и органы. Осложнения болезни приводят к смерти — как правило, продолжительность жизни с таким диагнозом не превышает 20 лет.
В основном заболевание наследуется по аутосомно-рецессивному типу. Это значит, что болезнь ребёнку передают здоровые родители — носители дефектного гена.
Читать статью целикомврач-эксперт



