
Фенилкетонурия


и делаем скидки до 30%
Начните экономить прямо сейчас!
Фенилкетонурия: общие сведения
Фенилкетонурия (ФКУ) — редкое генетическое заболевание, при котором организм не может нормально перерабатывать аминокислоту фенилаланин.
Фенилаланин — многофункциональное вещество, которое входит в состав белков, участвует в синтезе гормонов, поддерживает нервную систему и нормализует работу щитовидной железы.
В небольших количествах фенилаланин нужен организму, но при ФКУ он накапливается в крови и тканях и повреждает их. В первую очередь страдает головной мозг.
Фенилкетонурия относится к наследственным нарушениям обмена веществ: человек рождается с этой особенностью и живёт с ней всю жизнь. Болезнь считается редкой — в среднем она встречается у одного из нескольких тысяч новорождённых.
Несмотря на редкость, анализ на фенилкетонурию включили в обязательный неонатальный скринингНеонатальный скринингРанняя диагностика некоторых врождённых и наследственных заболеваний., который все младенцы проходят в первые дни жизни. Если болезнь выявить и начать лечить сразу, можно успешно контролировать её и предотвратить тяжёлое повреждение мозга и задержку умственного и физического развития.
Причины фенилкетонурии
Фенилкетонурия развивается из-за мутации в гене PAH, который отвечает за выработку фермента фенилаланингидроксилазы.
Этот фермент помогает организму перерабатывать аминокислоту фенилаланин, которая поступает с белковой пищей. Фермент расщепляет её и превращает в другое вещество — тирозин, которое нужно для работы мозга и синтеза гормонов.
Если фермента недостаточно, фенилаланин из пищи не может полноценно перерабатываться и начинает накапливаться в крови и тканях. Высокая концентрация его токсична для нервной системы и приводит к повреждению мозга.
Фенилкетонурия не связана с внешними факторами: её не могут спровоцировать вредные привычки или неблагоприятная экологическая обстановка. Это именно генетически обусловленное состояние.
Родители ребёнка с фенилкетонурией обычно здоровы и могут даже не знать, что в их генах есть отклонения.
Как наследуется фенилкетонурия
Фенилкетонурия наследуется по аутосомно‑рецессивному типу. Это означает, что ребёнок заболевает только в том случае, если получает по одному мутированному гену PAH от обоих родителей.
Каждый человек получает по одной копии гена от каждого из родителей. Поэтому у ребёнка всегда есть две копии гена, отвечающего за обмен фенилаланина: одна материнская и одна отцовская.
У родителей — носителей фенилкетонурии в каждом наборе генов есть одна здоровая копия PAH и одна с мутацией. Они сами не болеют, потому что одной здоровой копии достаточно для нормальной работы фермента. Но при зачатии каждый из них может передать ребёнку либо здоровую копию, либо мутированную.
Если ребёнку повезёт, он получит от родителей только одну мутированную копию гена. В этом случае за производство фермента будет отвечать вторая — здоровая, а он станет здоровым носителем, как и его родители. Если же он получит обе мутированные копии гена, то разовьётся фенилкетонурия.
Вероятность наследования фенилкетонурии от здоровых носителей:
- 25% — ребёнок унаследует два мутированных гена и заболеет фенилкетонурией;
- 50% — ребёнок получит один мутированный ген: будет здоров, но станет носителем, как родители;
- 25% — ребёнок получит два нормальных гена: будет здоров и не передаст мутацию своим детям.
Если один родитель болен фенилкетонурией (оба гена мутированные), а второй — носитель (один мутированный ген), риск рождения ребёнка с ФКУ около 50%.
При планировании беременности людям с фенилкетонурией рекомендуется консультация генетика и медико-генетическое обследование.
Как развивается фенилкетонурия
После рождения ребёнок начинает получать белок с грудным молоком или смесью, а вместе с ним — аминокислоту фенилаланин.
У младенца с ФКУ фермента, перерабатывающего фенилаланин, синтезируется слишком мало. Поэтому аминокислота начинает постепенно накапливаться в крови и в тканях, в первую очередь в головном мозге.
Высокая концентрация фенилаланина приводит к тому, что внутри мозга хуже формируются нервные связи и передаются сигналы. В результате умственное развитие замедляется, могут возникнуть судороги.
Если же выявить фенилкетонурию в ходе неонатального скрининга — в первые дни жизни ребёнка — и сразу начать лечение, то уровень фенилаланина обычно удаётся удерживать в допустимых пределах. В этом случае развитие ребёнка будет ближе к норме, а риск тяжёлых осложнений значительно снизится.
Симптомы фенилкетонурии
У новорождённых с фенилкетонурией обычно нет видимых признаков болезни — дети выглядят и ведут себя так же, как здоровые младенцы. Первые нарушения появляются по мере накопления фенилаланина в крови — как правило, это происходит в возрасте 2–6 месяцев.
Симптомы фенилкетонурии у младенцев и детей раннего возраста:
- задержка психомоторного развития: ребёнок позже начинает держать голову, сидеть, ползать, ходить;
- снижение активности: малыш меньше двигается, реже улыбается, медленнее реагирует на то, что происходит вокруг него;
- повышенная возбудимость или, наоборот, заторможенность: ребёнок хуже спит, много плачет, может стать вялым;
- судороги — внезапные подёргивания мышц, иногда с кратковременной потерей сознания: возникают примерно у 25–50% детей, не получающих лечение;
- специфический «затхлый» запах тела — напоминает плесень или мокрую шерсть: возникает из-за выделения избыточного фенилаланина с потом и мочой;
- светлая кожа, светлые волосы, голубые глаза: у детей с ФКУ часто снижена выработка пигмента меланина, который придаёт цвет коже, волосам и радужной оболочке глаз, потому что для синтеза пигмента также нужен фенилаланин;
- экзема и другие кожные высыпания: защитные функции кожи снижаются из-за нарушения обмена фенилаланина.
Если фенилкетонурию не лечить, к первому году жизни обычно становится заметна выраженная задержка развития: ребёнок позже начинает держать голову, сидеть, ползать, не интересуется окружающим, не реагирует на близких.
Со временем симптомы прогрессируют. В зависимости от формы фенилкетонурии, они могут довольно сильно различаться.
Симптомы у детей старшего возраста и взрослых:
- снижение интеллекта — от лёгких трудностей в обучении до тяжёлой умственной отсталости;
- нарушение поведения — гиперактивность, импульсивность, тревожность, депрессия;
- плохая память, сложности с концентрацией внимания;
- неспособность к планированию и самоконтролю;
- тремор (дрожание) рук, нарушение координации;
- судорожные приступы.
Чем выше уровень фенилаланина в крови и чем дольше он остаётся увеличенным, тем серьёзнее повреждение головного мозга. Даже если человек знает о болезни и получает лечение, но не до конца соблюдает рекомендации врача, могут появиться проблемы с памятью и концентрацией внимания, возможны перепады настроения, тревожность, а в тяжёлых случаях — судороги, тремор и нарушение координации движений.
Виды и стадии развития фенилкетонурии
В зависимости от степени нарушения выработки фермента фенилаланингидроксилазы и уровня фенилаланина в крови выделяют три основные формы фенилкетонурии.
Формы фенилкетонурии:
- лёгкая форма — фермент вырабатывается в небольшом количестве, поэтому фенилаланин частично перерабатывается. При такой форме тяжёлых симптомов обычно нет, строгая лечебная диета не требуется, но человеку следует регулярно наблюдаться у врача и следить за состоянием здоровья;
- умеренная форма — фермент вырабатывается в незначительном количестве, поэтому при этой форме уже нужна специальная диета, а также врач может назначить поддерживающую лекарственную терапию. Без лечения развивается задержка умственного развития;
- классическая форма — фермента либо нет совсем, либо его активность минимальна. Это самая тяжёлая форма болезни — при ней нужно пожизненно соблюдать строгую гипофенилаланиновую диету. Если болезнь не лечить, к первому году жизни развивается тяжёлая умственная отсталость, которая остаётся на всю жизнь.
Диагностика фенилкетонурии
Анализ на фенилкетонурию включён в обязательный неонатальный скрининг — его проходят все новорождённые в России начиная с 1993 года.
Исследование проводится на 3—5‑й день жизни ребёнка (у недоношенных — на 7—14‑й день). Наряду с фенилаланином по показаниям также определяют уровень тирозина — аминокислоты, которая в норме образуется из фенилаланина. При ФКУ фермента недостаточно, поэтому и уровень тирозина будет снижен. Для анализов у младенца берут каплю капиллярной крови из пятки, помещают её на специальный бланк и отправляют в лабораторию. Если уровень фенилаланина несколько повышен (более 120 мкмоль/л), дополнительно определяют соотношение фенилаланин/тирозин и также проводят повторно анализ на фенилаланин. Если в динамике он повышен существенно (более 240 мкмоль/л), а отношение фенилаланина к тирозину более 3, то родителям рекомендуют как можно скорее прийти на консультацию к генетику.
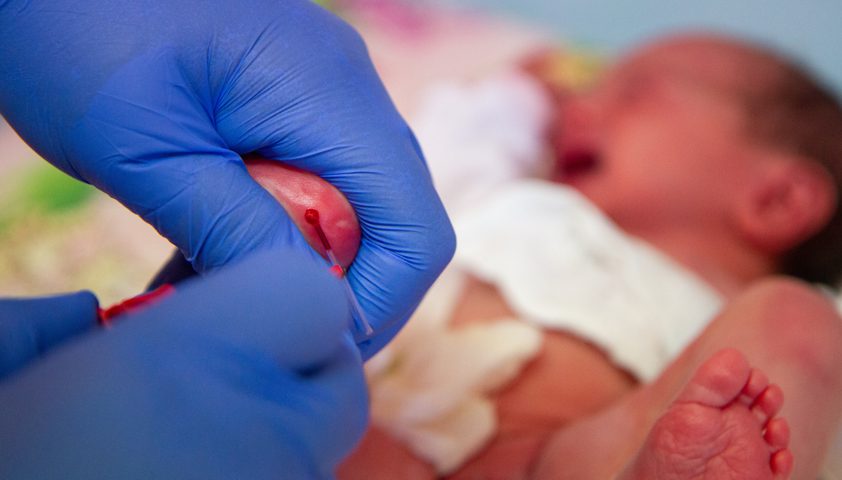
Анализ на ФКУ обычно берут сразу в роддоме
Благодаря неонатальному скринингу, ФКУ почти всегда выявляют в первые дни жизни ребёнка — ещё до появления симптомов. Это позволяет немедленно начать лечение и предотвратить повреждение головного мозга.
Если по какой-то причине младенцу не проводили скрининг или его результаты неизвестны, при подозрении на фенилкетонурию следует обратиться к неврологу. Врач проведёт осмотр, оценит умственное развитие и назначит необходимые обследования.
В первую очередь определяют концентрацию фенилаланина в крови. При ФКУ она может быть повышена в десятки раз.
Для подтверждения мутаций делают генетический анализ мутаций в гене PAH.
Могут потребоваться и инструментальные исследования. Например, МРТ (магнитно-резонансную томографию) головного мозга проводят, если есть неврологическая симптоматика, чтобы оценить степень структурного повреждения нервной ткани, выявить признаки повреждения оболочек нервных волокон, возможные очаговые изменения, особенно в областях, отвечающих за умственное развитие.
Лечение фенилкетонурии
Фенилкетонурию нельзя вылечить полностью, но её можно успешно контролировать. При правильной терапии дети с фенилкетонурией практически не уступают по развитию сверстникам: они могут учиться в обычной школе, строить карьеру и планировать семью.
Диета при фенилкетонурии
Основу лечения составляет пожизненная диета, ограничивающая поступление фенилаланина с пищей. Она позволяет поддерживать уровень аминокислоты в безопасных пределах и помогает защитить головной мозг от повреждения.
Фенилаланин входит в состав всех белков, поэтому лечебная диета получается довольно строгой. Основная цель — ограничить поступление фенилаланина с пищей до безопасного уровня, при этом обеспечив организм всеми необходимыми питательными веществами.
Запрещённые продукты:
- мясо и субпродукты — говядина, свинина, баранина, печень, почки, язык;
- птица — курица, индейка, утка, гусь;
- рыба и морепродукты — любая рыба, креветки, крабы, мидии;
- мясные и рыбные полуфабрикаты — колбасы, сосиски, сардельки, котлеты, консервы;
- яйца — куриные и перепелиные яйца в любом виде;
- молочные продукты с высоким содержанием белка — творог, сыры, сметана, йогурты, кефир, молоко;
- бобовые — фасоль, горох, соя, чечевица, нут;
- орехи и семена — все виды орехов, арахис, семена подсолнечника, кунжут;
- шоколад и какао — тёмный и молочный шоколад, какао-порошок, кондитерские изделия с какао;
- мучные изделия — хлеб, выпечка, блины, макароны из обычной пшеничной муки;
- крупы с высоким содержанием белка — гречка, овёс, пшено, перловка, манка;
- диетические напитки, сладости с аспартамом — этот подсластитель также содержит фенилаланин;
- жевательная резинка без сахара (часто содержит аспартам).
Помимо продуктов, запрещены некоторые лекарства в виде порошков и таблеток, потому что они зачастую содержат аспартам.

Людям с фенилкетонурией следует внимательно изучать состав диетических напитков и сладостей, жевательной резинки и даже лекарств
Разрешённые продукты:
- овощи — картофель, морковь, капуста (белокочанная, цветная, брокколи), огурцы, помидоры, кабачки, тыква, свёкла, репа, редис;
- фрукты и ягоды — яблоки, груши, виноград, дыня, арбуз, персики, абрикосы, сливы, вишня, клубника, малина, смородина, крыжовник;
- растительные масла — подсолнечное, оливковое, кукурузное, льняное масло;
- сахар и мёд — в умеренных количествах;
- специи и травы — соль, перец, лавровый лист, укроп, петрушка, базилик, корица, ванилин;
- крупы с низким содержанием белка — рис, кукурузная крупа, кукурузные хлопья;
- специализированные низкобелковые продукты для людей с фенилкетонурией — хлеб, макароны, мука, каши;
- фруктовые соки — натуральные, свежевыжатые;
- чай и кофе — без молока и добавок, содержащих аспартам;
- фруктовое мороженое — без молока и молочных компонентов.
Чтобы добрать норму белка, людям с фенилкетонурией нужно принимать специальные аминокислотные смеси — они содержат все необходимые аминокислоты, кроме фенилаланина. Смеси должны быть в рационе ежедневно и пожизненно — без них организм не получит необходимые вещества для роста и развития.
Младенцам разрешено грудное молоко в строго ограниченном количестве (его рассчитывает врач), а также специальные лечебные смеси без фенилаланина или с минимальным его содержанием.
Контроль уровня фенилаланина в крови
Обязательная часть лечения — постоянный мониторинг уровня фенилаланина в крови. Без анализов невозможно определить, правильно ли составлен рацион.
Периодичность исследований зависит от возраста: младенцам до 1 года анализ проводят 1–2 раза в неделю, детям от 1 до 12 лет — 1–4 раза в месяц, а подросткам и взрослым при стабильном уровне аминокислоты достаточно 1 раза в 1–3 месяца.
Целевой уровень фенилаланина в крови:
- 120–360 мкмоль/л (2–6 мг/дл) — для младенцев и детей до 12 лет;
- 120–600 мкмоль/л (2–10 мг/дл) — для подростков и взрослых;
- 120–360 мкмоль/л — для беременных.
У людей с фенилкетонурией важно проверять уровни витаминов и минералов, особенно железа, кальция, витамина D и B12. Также регулярно отслеживают показатели физического и умственного развития — рост и вес, речь, моторику.
Медикаментозное лечение
В дополнение к диете врач может назначить лекарственные препараты. Они помогают организму лучше перерабатывать фенилаланин и удерживать его уровень в крови в рамках нормальных значений. Благодаря этому части пациентов удаётся немного смягчить ограничения в питании и расширить рацион.
Например, врач может назначить препарат, который помогает ферменту работать активнее. Он подходит не всем пациентам (только в 20–30% случаев), поэтому его назначают после специального теста на чувствительность к препарату.
При огрехах в диете — если пациент случайно съел продукт с высоким содержанием белка, — врач может назначить инъекции с ферментом, который расщепляет фенилаланин в крови, быстро снижая его уровень. Такие инъекции назначаются только как экстренная мера — на случай единичного нарушения диеты.
Даже при приёме препаратов важно продолжать соблюдать диету.
Контроль фенилкетонурии во время беременности
Во время беременности особенно важно контролировать уровень фенилаланина в крови, потому что высокая концентрация этой аминокислоты токсична для плода.
Уровень фенилаланина не должен быть выше 120–360 мкмоль/л — у беременных его следует проверять 1–2 раза в неделю.
Если показатели выходят за рамки нормы, то фенилаланин повреждает плод, вызывая материнскую фенилкетонурию — это состояние, когда здоровый плод (у которого с генами всё в порядке) получает токсичное воздействие от матери. Это может привести к порокам сердца, задержке физического развития, микроцефалии (уменьшению размера головного мозга) и умственной отсталости.
Контролировать уровень фенилаланина важно уже на этапе планирования беременности.
Осложнения фенилкетонурии
Главная опасность фенилкетонурии — необратимое повреждение головного мозга, которое может привести к глубокой умственной отсталости. При классической форме фенилкетонурии без лечения она развивается уже к 1 году и остаётся на всю жизнь.
У состояния может быть множество других осложнений: они появляются, если человек не соблюдает рекомендации врача, например нарушает диету или и вовсе прекращает лечение — даже во взрослом возрасте.
Возможные осложнения ФКУ:
- необратимое повреждение головного мозга, которое приводит к тяжёлой умственной отсталости и задержке развития;
- низкорослость;
- избыточная масса тела и ожирение;
- микроцефалия (уменьшение окружности головы), позднее прорезывание зубов — нарушение физического развития;
- остеопения — снижение плотности костей;
- когнитивные нарушения — человек хуже усваивает и запоминает информацию, не может нормально сосредоточиться;
- снижение IQ при несоблюдении диеты даже во взрослом возрасте;
- микроцефалия плода (врождённое недоразвитие головного мозга), врождённые пороки сердца и другие нарушения;
- психические и эмоциональные нарушения — высокий уровень тревожности, эмоциональная неустойчивость, гиперактивность, агрессивность, депрессия, фобии, панические атаки.
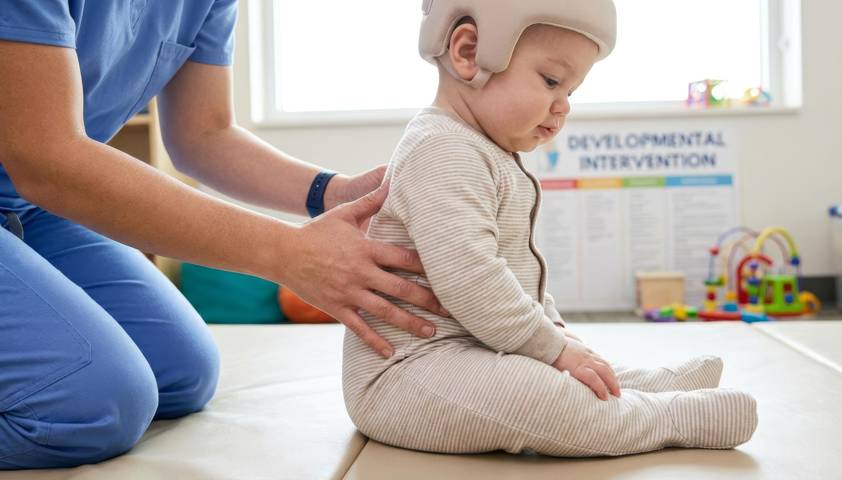
Ребёнок может так и не научиться говорить, узнавать близких, самостоятельно обслуживать себя
Из-за строгого ограничения белка в рационе может развиться дефицит питательных веществ: железа, кальция, витамина D, витамина B12. Это вредит развитию костей, крови и нервной системы.
Прогноз и профилактика фенилкетонурии
Если лечение начинают с первых дней жизни, прогноз обычно благоприятный. Дети с фенилкетонурией развиваются практически так же, как их здоровые сверстники: учатся в обычной школе, получают высшее образование, строят карьеру, создают семью.

Фенилкетонурия не влияет на продолжительность жизни, если человек контролирует уровень фенилаланина
Однако фенилкетонурия остаётся на всю жизнь. Даже если человек чувствует себя хорошо и не замечает симптомов, ему следует соблюдать диету и регулярно контролировать показатели.
Фенилкетонурия — генетическое заболевание, поэтому гарантированно предотвратить его появление невозможно. Однако родители могут оценить риски ещё до зачатия — для этого нужно обратиться к генетику и пройти обследование.
Особенно рекомендовано генетическое консультирование парам, у которых уже есть ребёнок фенилкетонурией, или если один из будущих родителей болен ФКУ.
Источники
- Классическая фенилкетонурия и другие виды гиперфенилаланинемии : клинические рекомендации / Союз педиатров России [и др.]. 2024.
- Классическая фенилкетонурия и другие виды гиперфенилаланинемии : клинические рекомендации / Союз педиатров России, Ассоциация медицинских генетиков, Ассоциация «Здоровье детей». 2024.
- Волгина С. Я., Яфарова С. Ш., Клетенкова Г. Р. Фенилкетонурия у детей: современные аспекты патогенеза, клинических проявлений, лечения // Российский вестник перинатологии и педиатрии. 2017. Т. 62. №5. С. 111–118.
- Боровик Т. Э., Ладодо К. С. Диетотерапия при классической фенилкетонурии: критерии выбора специализированных продуктов без фенилаланина // Вопросы современной педиатрии. 2013. Т. 12. №5. С. 40–48.
Частые вопросы
врач-эксперт



