
Синдром Марфана

и делаем скидки до 30%
Начните экономить прямо сейчас!
Синдром Марфана: общие сведения
Синдром Марфана — наследственное системное заболевание соединительной ткани, которое сопровождается поражением сердечно-сосудистой системы, опорно-двигательного аппарата, органов зрения.
Распространённость синдрома составляет примерно 1 случай на 5–10 тыс. человек. Болезнь одинаково часто встречается у мужчин и женщин и не зависит от этнической принадлежности.
Первым синдром описал в 1896 году французский педиатр Антуан Марфан, наблюдавший девочку с необычайно длинными руками и ногами, выраженными скелетными аномалиями. Позже стало очевидно, что заболевание затрагивает не только кости, но и сердце, крупные кровеносные сосуды и другие органы.
Причины синдрома Марфана
Основная причина синдрома Марфана — мутации в гене FBN1, который отвечает за синтез белка фибриллина-1, важнейшего компонента соединительной ткани.
Фибриллин-1 входит в состав микрофибрилл — тонких белковых нитей, входящих в состав кожи, связок, стенок кровеносных сосудов, лёгких и других органов.
Микрофибриллы образуют эластические волокна, благодаря которым ткани могут растягиваться, а затем возвращаться к своей первоначальной форме.
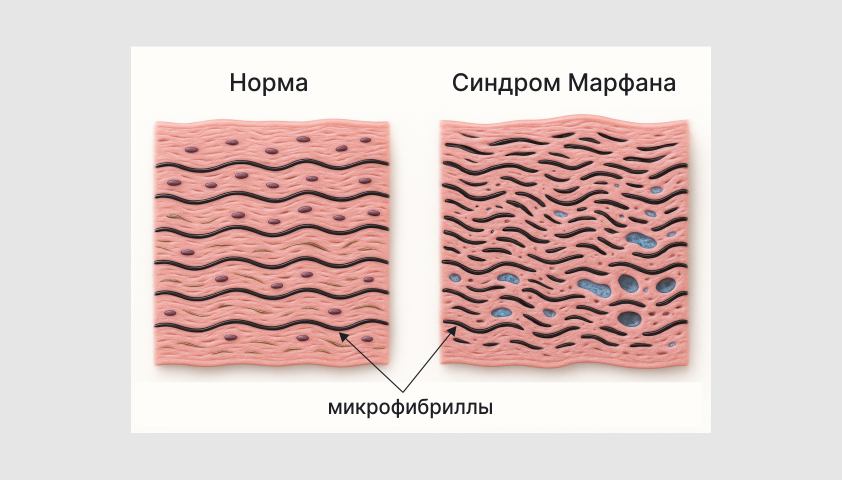
Схематичное изображение стенки кровеносного сосуда в норме и при синдроме Марфана
Из-за нарушения структуры фибриллина-1 микрофибриллы становятся дефектными, а соединительная ткань — менее прочной и устойчивой к механическим нагрузкам. У человека при этом формируется характерный набор внешних признаков: высокий рост, непропорционально длинные ноги и руки, длинные тонкие пальцы, деформации грудной клетки, искривление позвоночника, повышенная подвижность суставов.
Однако внешние особенности — лишь верхушка айсберга. Гораздо большее значение имеют изменения со стороны сердечно-сосудистой системы, прежде всего расширение аорты — крупнейшей артерии, по которой кровь от сердца поступает ко всем органам и тканям.
Чаще всего синдром Марфана наследуется по аутосомно-доминантному типу. Это значит, что если заболевание есть у одного из родителей, то вероятность его развития у ребёнка — 50%. Также болезнь может возникнуть спонтанно — из-за случайной генетической мутации.
Виды синдрома Марфана
В Международной классификации болезней и проблем, связанных со здоровьем, 10-го пересмотра (МКБ-10) синдрому Марфана присвоен код Q87.4.
Согласно МКБ, различают две основные формы синдрома Марфана.
При стёртой форме изменения затрагивают одну-две системы организма и часто остаются малозаметными. При выраженной — болезнь либо захватывает сразу несколько органов и систем, либо только одну, но повреждение при этом значительное.
Также условно выделяют три степени тяжести синдрома Марфана. При лёгкой степени симптомы неяркие. При среднетяжёлой возникают проблемы с позвоночником, глазами, сердцем, сосудами. А при тяжёлой происходит серьёзное повреждение внутренних органов, прежде всего аорты и сердечных клапанов.
Синдром Марфана может протекать по-разному. При классической форме у человека присутствует большинство характерных признаков заболевания и поражаются несколько органов и систем. При неполной, или мягкой, форме наблюдаются лишь отдельные проявления болезни, которые могут быть выражены незначительно.
Очень редкая форма — неонатальный синдром Марфана. Она сопровождается серьёзными нарушениями развития скелета, тяжёлым поражением сердца и быстрым развитием сердечной недостаточности в первые месяцы жизни ребёнка.
Симптомы синдрома Марфана
Клинические проявления синдрома Марфана чрезвычайно разнообразны.
Опорно-двигательный аппарат
Для людей, страдающих синдромом Марфана, характерна худощавость, высокий рост, непропорционально длинные руки и ноги, тонкие и длинные пальцы, гипермобильность суставов, сколиоз, кифоз, деформации грудной клетки по типу воронкообразной или килевидной, плоскостопие.
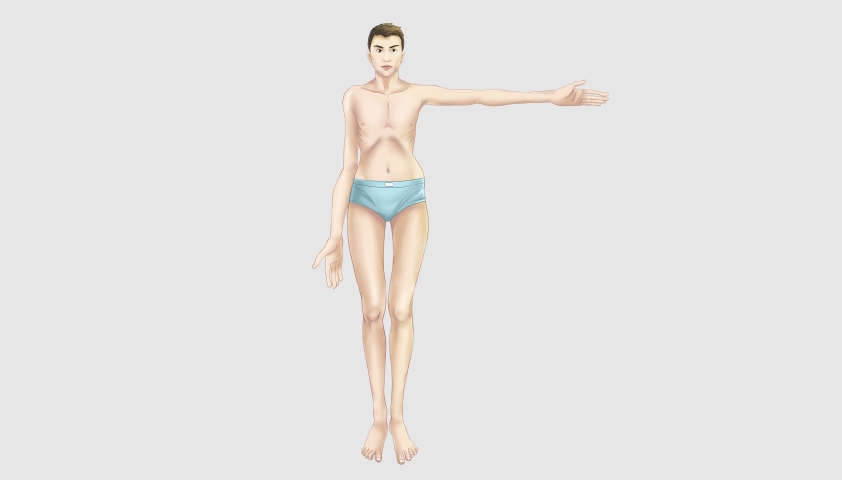
Так может выглядеть человек, страдающий синдромом Марфана
Зрение
Классическим проявлением синдрома Марфана считается эктопия (подвывих) хрусталика — смещение прозрачной линзы внутри глаза из-за слабости связок.
Кроме того, часто наблюдается выраженная близорукость, раннее развитие катаракты и глаукомы, повышенный риск отслойки сетчатки.
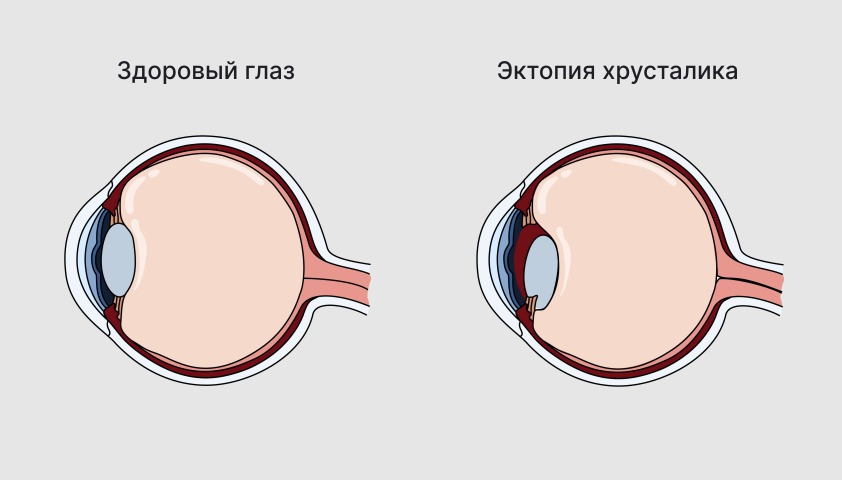
При синдроме Марфана связки, удерживающие хрусталик, растягиваются. Из-за этого он может частично или полностью сместиться
Сердечно-сосудистая система
Особое значение имеют сердечно-сосудистые проявления, поскольку именно они определяют прогноз заболевания. Наиболее типичное из них — прогрессирующее расширение корня аорты с формированием аневризмы.
Аорта — это полая трубка, по которой за жизнь в среднем проходит около 200 млн литров крови
Также может выявляться пролапс митрального клапанаПролапс митрального клапанаНеполное смыкание створок клапана сердца, из-за чего часть крови течёт обратно., недостаточность клапанов сердца, расширение его камер и нарушения сердечного ритма.
Без лечения именно повреждение сердца и крупных сосудов чаще всего становится причиной тяжёлых осложнений и преждевременной смерти.
Дыхательная система
У людей с синдромом Марфана часто обнаруживается апноэ (неоднократные остановки дыхания во сне) и воздушные полости — буллы — в лёгких.
Повышается риск спонтанного пневмоторакса — грозного состояния, при котором воздух попадает в полость вокруг лёгкого, сдавливая его и нарушая дыхание.
Кожа
У людей с синдромом Марфана часто появляются стрии (растяжки) — линейные дефекты кожи, возникающие из-за её внутреннего надрыва и последующего рубцевания, а также послеоперационные и паховые грыжи.
Неврологические нарушения
Для синдрома Марфана наиболее характерна дуральная эктазия — расширение твёрдой мозговой оболочки позвоночного канала, которое может сопровождаться хроническими болями в пояснице, головными болями, онемением, покалыванием и слабостью в ногах.
Осложнения синдрома Марфана
Главная опасность синдрома Марфана связана не с особенностями внешности или деформациями скелета, а с риском осложнений со стороны сердечно-сосудистой системы.
Наиболее тяжёлым из них считается расслоение аорты. Оно возникает из-за разрыва внутреннего слоя сосудистой стенки и проникновения крови между слоями аорты. Ключевые симптомы — внезапная сильная боль в груди или спине, резкое ухудшение самочувствия.
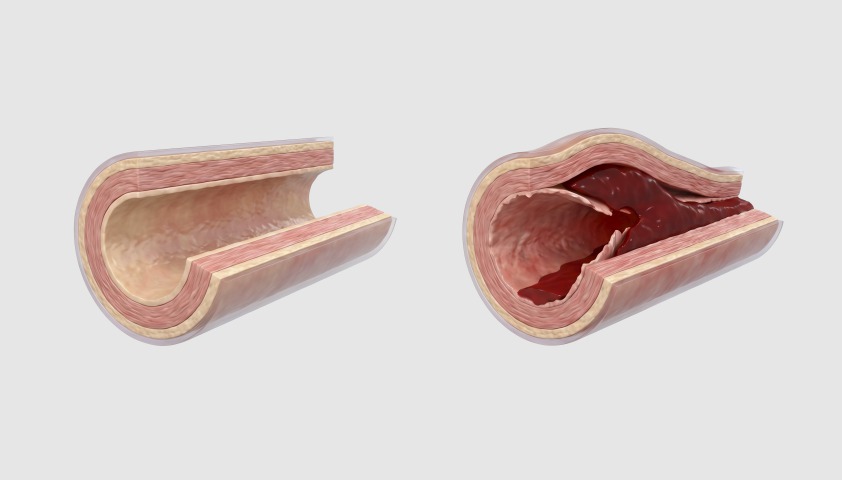
Слева — здоровый сосуд, справа — расслоение аорты
Не менее опасен разрыв аневризмы аорты, который требует экстренной операции.
К другим возможным осложнениям синдрома Марфана относится сердечная недостаточностьСердечная недостаточностьСостояние, при котором сердце не может перекачивать достаточное количество крови к тканям и органам., отслойка сетчатки, спонтанный пневмоторакс.
Диагностика синдрома Марфана
Диагностика синдрома Марфана основывается на пересмотренных Гентских критериях (Revised Ghent Criteria). При постановке диагноза учитываются такие признаки, как расширение корня аорты, эктопия хрусталика, патогенная мутация в гене FBN1 и совокупность системных признаков поражения соединительной ткани.
Гентские критерии не предназначены для самодиагностики. Оценивать их результаты и ставить диагноз может только врач.
Инструментальная диагностика
Один из основных методов обследования при синдроме Марфана — эхокардиография (УЗИ сердца), которая позволяет оценить размеры аорты, работу клапанов и состояние сердечной мышцы.
-
УЗИ сердцавт, 21 июл3 200 ₽
Для более подробного исследования аорты врач может назначить компьютерную (КТ) или магнитно-резонансную (МРТ) томографию.
Обследование у офтальмолога помогает выявить смещение хрусталика, близорукость и другие нарушения зрения. Для оценки изменений со стороны скелета используются рентгенография и другие методы визуализации.
Генетическая диагностика
Генетическое исследование помогает подтвердить диагноз «синдром Марфана» и отличить его от других наследственных заболеваний соединительной ткани.
Помимо гена FBN1, могут анализироваться гены TGFBR1, TGFBR2, SMAD3, TGFB2 и TGFB3, связанные с заболеваниями аорты и соединительной ткани. Это позволяет провести точную диагностику и подобрать правильную тактику наблюдения.
Лечение синдрома Марфана
Синдром Марфана — это наследственное заболевание, поэтому полностью вылечить его пока невозможно. Основная задача терапии — предотвратить осложнения и замедлить изменения в аорте.
Для этого назначают лекарства, которые снижают нагрузку на сердце и стенки аорты. Чаще всего используются бета-адреноблокаторы и лозартан.
Если аорта значительно расширена или существует высокий риск её расслоения, может потребоваться операция. Современные хирургические методы позволяют укрепить или заменить поражённый участок аорты, а также скорректировать пороки клапанов сердца.
Учёные продолжают разрабатывать новые методы терапии. Одним из самых перспективных направлений считается генное редактирование, которое в будущем может позволить исправлять генетические мутации. Однако пока эта технология не применяется в клинической практике.
Профилактика осложнений синдрома Марфана
Чтобы снизить риск осложнений, людям с синдромом Марфана важно регулярно проходить обследования. Рекомендуется ежегодно проверять состояние аорты и контролировать артериальное давление. Следует избегать тяжёлых физических нагрузок и силовых видов спорта.
При необходимости врачи могут рекомендовать операцию на аорте, чтобы предотвратить опасные осложнения. Также важно регулярно посещать офтальмолога и пройти генетическое консультирование, особенно если в семье есть случаи заболевания.
Пациентам с синдромом Марфана рекомендуется пожизненное наблюдение у кардиолога и других специалистов.
Во время беременности нагрузка на сердце и сосуды увеличивается, поэтому у женщин с синдромом Марфана возрастает риск осложнений со стороны аорты. По этой причине беременность должна проходить под тщательным наблюдением врачей. Будущей маме важно регулярно посещать кардиолога, проходить обследования сердца и аорты, а также получить генетическую консультацию, чтобы оценить риск передачи заболевания ребёнку.
Прогноз синдрома Марфана
Прогноз синдрома Марфана значительно улучшился за последние десятилетия.
Если в прошлом средняя продолжительность жизни пациентов с таким диагнозом составляла около 30–40 лет, то благодаря ранней диагностике, современным методам медикаментозного лечения, профилактической хирургии и регулярному наблюдению продолжительность жизни большинства людей сегодня приближается к показателям общей популяции.
Источники
- Лунева Е. Б., Парфенова Н. Н., Коршунова А. Л. и др. Новые подходы к диагностике синдрома Марфана // Российский семейный врач. 2012. Т. 16. №3. С. 14–19.
- Румянцева В. А., Чарчян Э. Р., Заклязьминская Е. В. и др. Синдром Марфана, вызванный нонсенс-мутацией в гене фибриллина: клиническое применение ДНК-диагностики в хирургии аорты // Клиническая и экспериментальная хирургия. 2015. №2. С. 97–103.
- Pratt B., Curci J. Arterial elastic fiber structure, function and potential roles in acute aortic dissection // The Journal of Cardiovascular Surgery. 2010. Vol. 51(5). P. 647–656.
Частые вопросы
врач-эксперт



